

The Cotney Lab is broadly interested in determining how gene regulatory elements, namely enhancers, control gene expression during mammalian development. We aim to understand how new gene regulatory functions evolve, to identify mechanisms of enhancer function over large genomic distances, and globally identify variants of enhancer sequences that are associated with human disease. Understanding how enhancers participate in such processes and the regulatory information contained within them will provide new avenues for dissecting gene regulation during development and lead to better prediction of disease risk from primary genetic sequence.
Evolution of mammalian developmental enhancers
For over 30 years researchers have hypothesized that the small number of differences in protein-coding genes between closely related mammalian species (i.e. human vs chimp) are likely not sufficient to explain significant differences in physiology and morphology. Many have postulated that species differences in form and function likely arise not from differences in the functions of genes themselves but how their expression is regulated in both the adult and developing embryo (King and Wilson, 1975). Dr. Cotney addressed this hypothesis during his postdoctoral work in the Noonan lab through comparative epigenetic analysis of human, rhesus monkey and mouse limb development (Cotney et al., 2013). He developed experimental techniques to perform global profiling of activation-associated post-translational modifications of histone proteins in extremely small amounts of embryonic tissue and computational pipelines to process and compare these genomic signals across multiple genomes. These techniques allowed the identification of thousands of sequences that are potentially regulating gene expression in a defined human tissue and developmental timeframe that are distinct from activities observed in the homologous developmental states of rhesus monkeys and mice. Along with other members of the Noonan lab, we then integrated these data with global limb transcriptome data and human population data to identify regulatory changes likely to have biological function in human limb development. The human gains of activation based on histone modification levels are significantly associated with increased expression of approximately 300 genes in human limb tissue compared to similar stages in mouse suggesting bona fide regulatory change in embryonic limb development. This work also demonstrated the first in vivo experimental evidence of a gain of enhancer function in human embryonic development and provides an initial estimate for the rate of gene regulatory change in embryonic development.
However, it remains to be determined if this trend is the same across similar evolutionary time scales and multiple developmental tissues. The Cotney Lab aims to employ these comparative functional genomics techniques in multiple rodent species that span evolutionary distances similar to those estimated between human and other apes or old world monkeys (up to 25 million years). We will comprehensively identify enhancers and genes active during developmental patterning of several structures (i.e. brain, lung, heart, and limb) in several rodent species using multiple functional marks. We will then compare the activation states, target gene expression levels, and sequence composition of these enhancers both within and across species. Using inbred rodent species for these types of experiments compared to the previous studies has several key advantages. First, examining closely related laboratory strains of C57BL6/J and A/J mice to other mouse species and the Norwegian Rat will allow interrogation of well defined evolutionary distances and reduce impacts of inter-individual genetic variability. Second, they have similar lengths of gestation and similar developmental morphology that allow unambiguous dissection of homologous tissues across species. Third, embryos can be generated at a scale that cannot be matched by timed rhesus pregnancies and collection of donated human embryonic material. Lastly, the function of a particular enhancer in multiple genetic backgrounds can be assessed during embryonic development.
Related to this last concept we aim to identify the functional sequence changes in developmental enhancers that impart species-specific function on a global scale. We will utilize previously published high-throughput enhancer readout assays, STARR-Seq (Arnold et al., 2013), in a genome-wide fashion to compare enhancer activity from several species. Briefly, the entire genome of a species is sheared to ~500 bp and inserted downstream of a minimal promoter and reporter gene. Instead of directing expression of only a reporter gene, the enhancer sequence is also transcribed producing a direct readout of enhancer activity in RNA form. This will allow enhancer activity comparisons in a common trans-environment that models a developmental state or tissue, such as limb derived mouse embryonic fibroblasts or neural crest cells differentiated from embryonic stem cells. Identifying the rate at which developmental enhancer sequence content changes and ultimately how their gene regulatory capacity is altered will deepen our understanding of the evolutionary forces controlling embryonic development, identify commonly targeted genetic architecture, and begin to unravel the complex information encoded within these regulatory sequences.
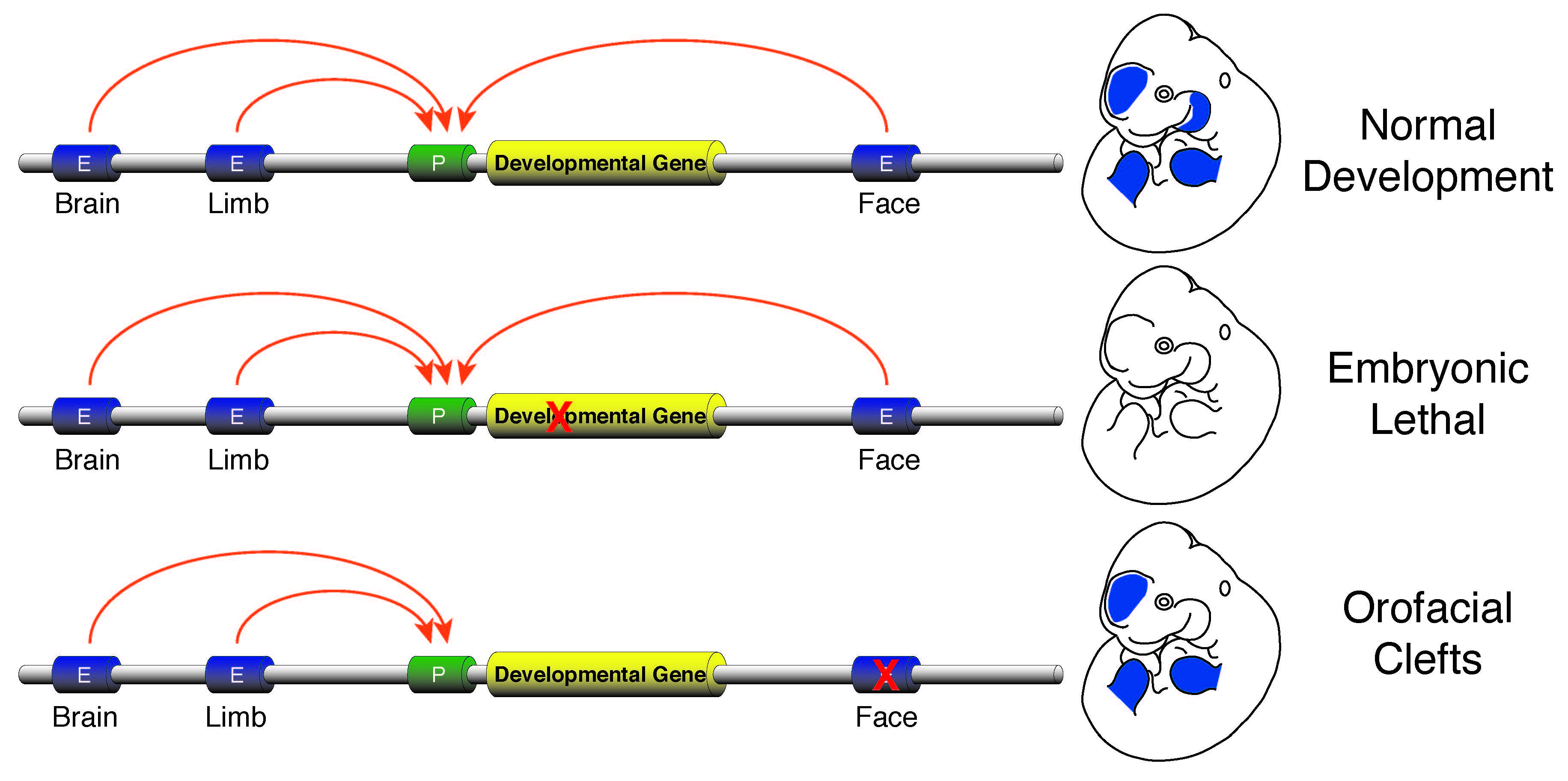
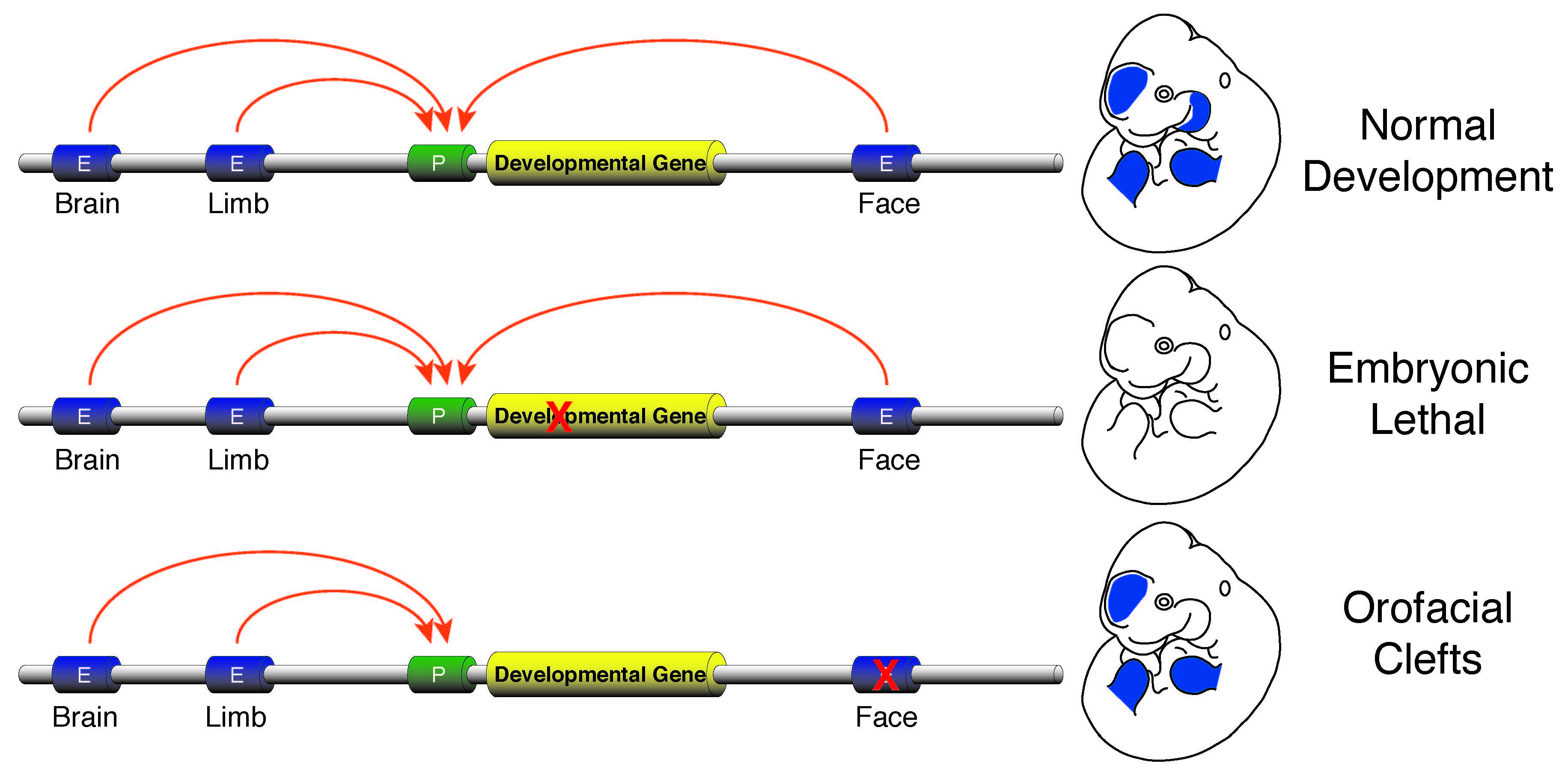
Enhancer sequence variation and human disease
The vast majority of commonly occurring variants identified by genome-wide association studies for a variety of diseases are located in non-coding portions of the genome suggesting improper regulation of gene expression is a major contributor to human disease (Rivera and Ren, 2013). I plan investigate this issue by developing a research program focused on identification of causal enhancer variation in developmental defects and begin to unravel how commonly occurring variants interact with a specific genetic background to produce a disease phenotype. Genes required for developmental patterning, such as homeobox transcription factors or signaling molecules like Sonic hedgehog (SHH), have complex expression patterns in multiple embryonic tissues. Activation of such genes in a precise spatiotemporal fashion is facilitated by numerous enhancers found in the non-genic portions of the genome. Loss of function mutations in genes involved in patterning are typically lethal or cause gross abnormalities, while disruptions of the enhancers that control those genes are much less dramatic and isolated to specific tissues. The tissue-specific nature of the few known enhancer defects make non-syndromic birth defects an area ripe for investigation of the general role of enhancer dysfunction and human disease. I aim to initially leverage commonly occurring non-syndromic cleft lip and palate (NSCLP) defects to identify causal enhancer variation and dissect the mechanisms that are affected in these disorders.
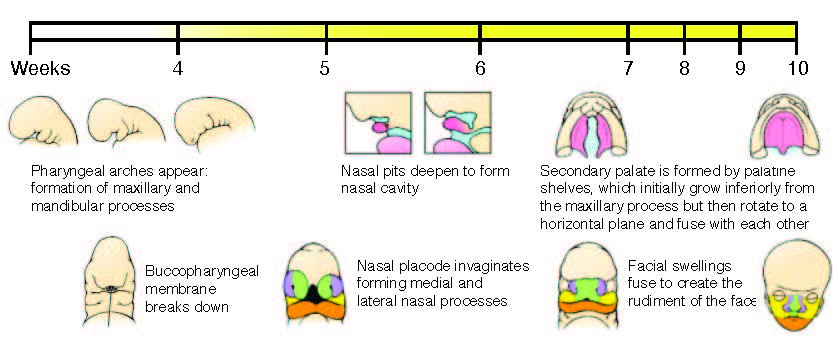
Figure adapted from Schoenwolf, G.C., Bleyl, S.B., Brauer, P.R., and Francis-West, P.H. (2009). Larsen’s Human Embryology (Ann Arbor, MI: Churchill Livingstone/Elsevier).
Human orofacial clefts can be diagnosed at approximately 16 weeks of gestation (Costello et al., 2008), however they occur as early as 5 weeks (Diewert, 1985; Schoenwolf et al., 2009). Therefore, to identify processes that are disrupted by variants in regulatory sequences, tissue from these early stages must be interrogated when important regulatory pathways are active. Using experimental and computational methods Dr. Cotney developed to globally profile active promoters and enhancers in small amounts of embryonic tissue (Cotney et al., 2013; 2012), we will globally characterize craniofacial gene regulation directly in human embryonic tissue at the developmental stages when fusion of orofacial structures is determined. We will integrate active enhancer and promoter maps with NSCLP associated SNPs to identify regulatory elements that potentially influence the formation of NSCLP. We will screen enhancers in strong linkage disequilibrium with NSCLP variants in a patient cohort to identify candidate causative variation using molecular inversion probe technology (O’Roak et al., 2012). We then aim to functionally characterize the regulatory capacity of the identified enhancers and NSCLP associated alleles in mouse craniofacial embryonic development. These experiments will identify the stages of development that these enhancers exert regulatory control, will begin to reveal the genetic architecture of NSCLP, and provide targets for future genetic screening. This line of experimental work will establish a paradigm for understanding virtually any developmental disorder that is caused, at least in part, by improper gene regulation during embryonic patterning.
 Long-distance regulation of gene expression
Long-distance regulation of gene expression
To fully understand how developmental enhancers influence phenotype and cause disease their regulatory targets must be identified. Enhancers can be found in close proximity to their regulatory targets or potentially operate over entire chromosomes. Recent studies indicate chromosomes maintain static topological domains that are several megabases in size that limit the scope of enhancer influence and establish microcosms of co-regulated genes (Dixon et al., 2012). Within these large topological domains, enhancers have been shown to interact with target promoters through poorly understood looping mechanisms (Sanyal et al., 2012). The majority of loops that have been detected thus far are stable across tissues, developmental contexts, and species (Demare et al., 2013; Handoko et al., 2011; Phillips-Cremins et al., 2013). This could indicate the looping events are static and are not the drivers of gene activation as many models have suggested. Alternatively current methods are only capable of detecting the most stable or frequent events and many enhancer-promoter interactions are dynamic and only occur in specific contexts. Histone modification data and the specificity of many experimentally tested developmental enhancers would suggest that the latter is a more likely scenario but this remains to be determined. We aim to identify the targets of developmental enhancers and determine the mechanism by which tissue-specific developmental enhancers identify and interact with these targets. This will require refinement of current chromosome conformation capture experiments and application to developmental contexts, such as defined stages of mammalian limb development, where many tissue specific enhancers and likely targets have been previously identified. We will leverage experience from the design and implementation of the first ChIA-PET experiment in embryonic tissue (Demare et al., 2013) to refine methods for identifying promoter-enhancer and enhancer-enhancer interactions that regulate gene expression in a tissue-specific fashion. We have identified a number of limb-specific regulatory domains in the mouse that can be interrogated to identify dynamics of interactions and the proteins that are found in these tissue-specific complexes (Cotney et al., 2012; 2013). Identifying how enhancers and promoters interact in a tissue-specific fashion and the proteins facilitating these interactions will increase our understanding of precisely controlled spatiotemporal gene expression during embryonic development. This will provide a basis to dissect developmental disorders that are likely to arise from improperly controlled gene expression.